By Prof. Dr. Christian Haass, Laboratory for Alzheimer’s and Parkinson’s Research, Chair of Metabolic Biochemistry at Ludwig Maximilian University, Munich
Almost exactly 100 years ago, a patient came to the practice of the Munich psychiatrist Dr. Alois Alzheimer, who described her disease with the following harrowing words: „I have lost myself“. This patient was to be the world’s first Alzheimer’s case and is known today as the famous case of „Auguste D.“ Of course, there were many other cases of senile forgetfulness even back then, as it is also very aptly depicted artistically in Paul Klee’s „The Forgetful Angel.“ Today, Alzheimer’s disease is the most common dementia worldwide. In Germany alone, almost 1.2 million Alzheimer’s patients are currently known. Since our average age has massively increased (and will continue to increase) due to enormous medical progress, we must expect a veritable flood of Alzheimer’s cases.
Old people’s homes, nursing homes and health insurance companies are not prepared for this problem. The development of an effective therapy against this „widespread disease“ is absolutely necessary. Current medications can only temporarily alleviate the symptoms a little, but they can in no way stop the creeping memory loss. To achieve this, a precise understanding of the molecular and cell biological mechanisms of action is necessary. So-called target molecules must be identified, the blocking (inhibition) of which slows down the course of the disease or even stops it completely.
To achieve this, we want to understand how the pathological deposition of the characteristic amyloid plaques occurs in the Alzheimer brain. Our hypothesis is that blocking plaque formation should stop the progression of the disease and perhaps even, if intervened in time, prevent it from occurring in the first place. Amyloid plaques are composed of a small protein called amyloid. The amyloid itself is apparently cut from a much larger protein, whereupon it clumps together, forms small aggregates, and eventually accumulates in the plaques. The small aggregates and the plaques themselves damage surrounding nerve cells so severely that they eventually die. The increasing loss of nerve cells over the years then leads to the dramatic loss of memory in patients.
While still an assistant professor at Harvard University in Boston, I made the very surprising discovery while studying simple human cells that the formation of amyloid is a completely normal process. It happens all the time in all of us from the day we are born. Unfortunately, however, this means that a time bomb is ticking inside us, which in old age, when enough amyloid has accumulated, leads to an explosion of plaques and nerve cell death. This fact teaches that the disease can and will indeed strike us all – if only we get old enough. In the lab, we are using this discovery to understand the precise mechanisms of how amyloid is cut out. To do this, we are working with human cellular models, yeast, and various in vivo systems, including genetically engineered (transgenic) lines.
Our goal has been to find the „scissor-like“ enzymes that cut out the toxic amyloid. We now refer to these scissors as „secretases.“ There is one typical secretase for each of the two cuts (the β-, and the γ-secretase), which we studied in detail to determine how they function. Using simple baker’s yeast, we found that the γ-secretase has a completely surprising structure. It actually resembles a conventional paper scissors in its structure, construction and functioning. We found two blades, each on two different subunits, held together by a screw and a nut. Using a modern genetic engineering method, we were able to specifically unscrew the screw. This resulted in the two cutting edges falling apart and being recognized and degraded by the cell as waste. The absence of the active γ-secretase then had quite massive consequences for the formation of the toxic amyloid. Its production was almost completely blocked, meaning that we had indeed identified the correct target molecule and prepared a basis for targeted drug development.
We are now using fully automated tests to look for small chemical compounds that specifically and selectively prevent the scissors from snapping shut. At the same time, we naturally have a great interest in finding out what normal functions the scissors have in our bodies. This is not only of enormous academic interest, but also plays a highly significant role in therapy, since we must not block essential functions in the human body under any circumstances. We have studied the biological function of scissors in simple model systems. For this purpose, we recently introduced zebrafish technology with the help of the Leibniz Prize of the DFG and the active support of the LMU. This system allowed us not only to identify the normal function of the scissors (in cell differentiation), but also to describe side effects of inhibitors of the scissors. Our model is therefore now ideally suited for targeting drugs that do not show any of the undesirable side effects. Furthermore, we are in the process of genetically manipulating the fish in such a way that it shows Alzheimer’s symptoms including memory loss (for example, a fish does remember in which of the four corners of an aquarium it gets its food). We will then modify the genome of this fish so that each gene is altered individually. To make this possible, we have set up a large fish house, which currently contains over 1000 aquariums.
The goal is to find artificial mutants in which the Alzheimer’s symptoms are either attenuated or intensified. By cloning the corresponding genes, we hope to gain new insights into previously completely unknown aspects of Alzheimer’s disease, into the mechanisms that lead to the death of nerve cells through amyloid aggregation.
Our work is carried out in local (SFB 596 Molecular Mechanisms of Alzheimer’s Disease), national (DFG, Focus Cell Biology of Alzheimer’s Disease), European (European Community Grants) and American (American Health Assistance Foundation) collaborative systems. There are active collaborations with pharmaceutical companies (Boehringer Ingelheim KG; Merck Sharp & Dome; Hofmann La Roche).
By Prof. Dr. Christian Haass, Laboratory for Alzheimer’s and Parkinson’s Disease Research, Chair of Metabolic Biochemistry at Ludwig-Maximilians-University, Munich.
Alzheimer’s disease is the most common form of dementia worldwide, and almost every reader of this article knows someone among his or her friends and relatives who is afflicted by this scourge of the 20th and 21st centuries. The increasingly frequent occurrence of Alzheimer’s disease is related to the dramatic increase in life expectancy. Unfortunately, the older we get, the higher the chance of developing the first symptoms. What is initially characterized by a minor forgetfulness, can in the final stage lead to a complete loss of personality – an unimaginable drama for those affected and their relatives. Due to the age-related increase in Alzheimer’s disease, we will have to reckon with a massive explosion in the number of patients with increasingly higher medical standards, a fact for which health politicians in Germany are not yet prepared. There is a lack of personnel, nursing homes and financial resources for the care of patients with dementia. There are no interdisciplinary research centers in Germany that deal specifically with the mechanisms of age-related diseases.
I was able to put together the first national network of German Alzheimer’s researchers only five years ago with the help of the German Research Foundation (DFG), an initiative that had already been started in the USA in the 1980s. This is all the more amazing because some of the most fundamental and exciting discoveries about the mechanisms of Alzheimer’s disease were made in Germany. I will only mention here the original discovery of the pathology by Alois Alzheimer (100 years ago), the cloning of the „Alzheimer’s gene“ and the identification of one of the central Alzheimer’s enzymes which I will discuss in more detail later.
In the following, I will first describe the pathology of the disease and then present the molecular mechanisms responsible for it. At the end, I will discuss new possibilities of therapy and the related hopes and difficulties.
Aggregating amyloids trigger a lethal cascade
Pathologically, Alzheimer’s disease is characterized, among other things, by the appearance of countless amyloid plaques in the brains of patients (Fig. 1). In some regions of the brain, these plaques can occupy up to 20% of the brain volume. While the plaques are located outside the neurons, additional clumps, which we call tangles, are found in numerous neurons (Fig. 1). In the affected regions of the brain, there is a dramatic loss of neurons. Amyloid plaques thus apparently contain a molecule that kills surrounding nerve cells. This molecule turned out to be a small protein that we now call amyloid ß-peptide (amyloid). Amyloid tends to aggregate with itself and then be deposited as an insoluble complab. Such aggregating cytotoxic peptides are a typical feature of many neurodegenerative diseases, with a specific amyloid molecule being formed in each disease. However, amyloids from a wide variety of diseases appear to have one thing in common: refolding of their natural three-dimensional structure promotes aggregation and consequent precipitation. Accumulation of these aggregates consisting of a wide variety of amyloids then appears to initiate neuronal cell death.
In the case of Alzheimer’s disease, small oligomeric amyloid aggregates (precursors to the actual plaques) trigger a lethal cascade, now known as the amyloid cascade. Among other things, this causes tangle formation (black „bundles“ in Fig.1 on the right), which in turn induce the formation of numerous toxic molecules, the consequence of which is then the death of nerve cells. In addition, the small amyloid oligomers also have a direct effect on our memory processes. Even the smallest traces can directly affect the manifestation of learned knowledge. Since amyloid triggers the amyloid cascade according to our current knowledge, this molecule is an ideal target for new, cause-oriented forms of therapy. Here, one searches for the genes and the enzymes encoded by them (so-called targets or target molecules) that are responsible for the formation of the amyloid molecule. This approach does not, of course, exclude other molecules further down the cascade (such as the tangles).
Familial Alzheimer’s disease
In a few cases (maximum 1-5%), Alzheimer’s disease occurs in a particularly aggressive form within individual families. This variant of the disease is pathologically indistinguishable from the large number of sporadic Alzheimer’s cases, but it breaks out very early. Cases are known in which a vehement onset of Alzheimer’s disease occurred at the age of less than 30 years. An accumulation of severe Alzheimer cases with early onset in individual families therefore already implies that genetic risk factors are present here, which are passed on. These risk factors, if appropriately altered by mutations, seem to dramatically accelerate the course of Alzheimer’s pathology and especially amyloid plaque formation. For molecular biologists, therefore, these genes hold the key to understanding the cellular mechanisms of the disease, although of course the number of genetically inherited AD cases is epidemiologically irrelevant.
In the following, mutations in three genes that cause Alzheimer’s disease will be described, and at the end recent research results will be presented that show that inactivation of certain Alzheimer’s genes leads to a massive reduction of amyloid formation. Mutations in three genes accelerate the aggregation of the amyloid molecule.
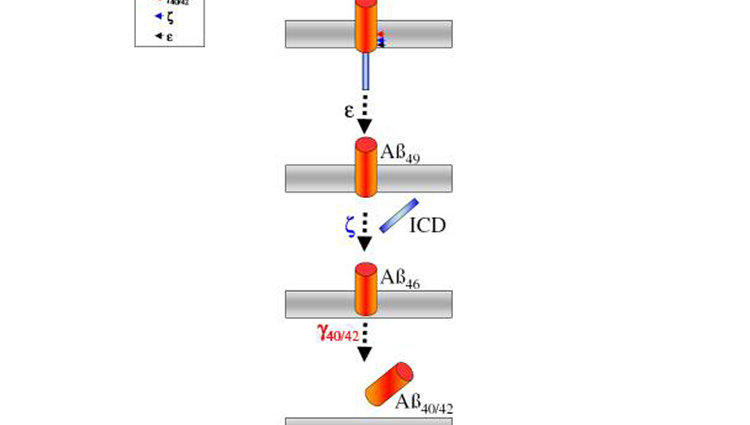
The first mutations were found in the gene of the amyloid precursor itself. These mutations are not scattered throughout the ßAPP molecule, but occur exactly at the interfaces of the three secretases. Ultimately, all these mutations have one and the same effect: they cause the production of a minimally modified amyloid molecule. In contrast to the 40 building blocks (amino acids) long Aß that we all constantly produce, the mutations increase the formation of an Aß molecule elongated by only two building blocks (Fig. 4). This variant has the fatal property of aggregating extremely rapidly with itself (Fig. 4). The mutations therefore reduce the time normally required for amyloid plaque formation to a minimum, leading to very early onset of the disease, but without altering the characteristic pathology as such.
Much more frequent mutations have been found in the two very similar presenil genes, (abbreviated PS1 and PS2). All mutations studied so far, just like the mutations in the amyloid precursor, cause an increased formation of the amyloid molecule elongated by two amino acids (Fig. 4). This again leads to accelerated formation of Alzheimer’s pathology, which can then occur as early as the second decade of life. In nature, changes thus occur in genes that quite obviously (as in the case of the precursor gene) directly influence amyloid formation. However, the much more frequent mutations in the presenilins were very difficult to explain. What do the presenilins have to do with amyloid formation? How do these influence amyloid formation?
Much more frequent mutations were found in the two very similar presenilins, (abbreviated PS1 and PS2). All the mutations studied so far, just like the mutations in the amyloid precursor, cause increased formation of the amyloid molecule, which is elongated by two amino acids (Fig. 4). This again leads to accelerated formation of Alzheimer’s pathology, which can then occur as early as the second decade of life. In nature, changes thus occur in genes that quite obviously (as in the case of the precursor gene) directly influence amyloid formation. The much The presenilins are an important building block of g-secretase.
For decades, countless laboratories have been searching for g-secretase as the key enzyme in Alzheimer’s disease, sometimes in very elaborate (but nevertheless unsuccessful) experiments. As always, the key lay in a conceivably simple conclusion. Let us take another look at the effect the mutations in the presenilin genes have on amyloid formation (Fig. 4). Well over 100 different mutations distributed over the entire gene all have, strangely enough, one and the same effect: they shift the cut of the g-secretase by exactly 2 building blocks, resulting in a longer, faster aggregating amyloid. So how can these mutations affect g-secretase so fatally? The solution was very simple: the presenilins are identical to the g-secretase, or at least an important building block of the g-secretase!
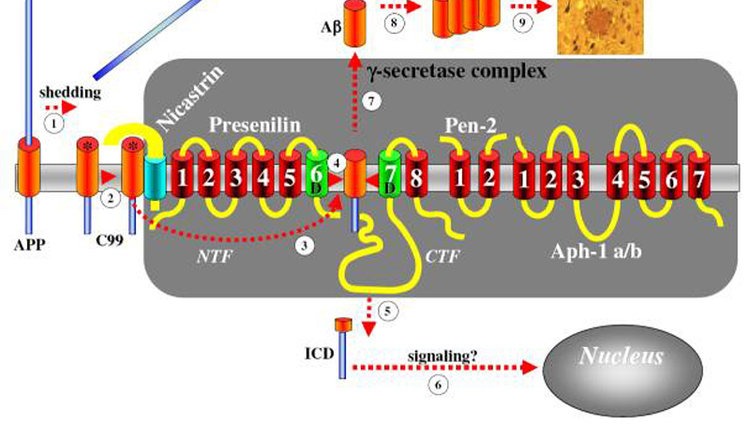
To understand the structure of g-secretase, we should have the image of a typical pair of paper scissors in mind (Fig. 2). These consist of two blades that perform the actual cutting process. The blades are identical to the presenilines. These are always in the form of two separate parts, both of which are absolutely necessary for the cutting process. However, the separated blades must also be held together. For this purpose, screws and nuts are used in mechanics (Fig. 2). This is exactly what evolution has „copied“. Two additional genes (or their protein products) are needed to build the scissors and hold them together. The screw is held together by a protein called nicastrin and dzu. The screw is formed by a protein called nicastrin and the nut by Pen-2 (Fig. 2). Both together hold the blades of the secretase together and allow their precise snapping shut.
Our current understanding of g-secretase (and thus of amyloid formation in general) would be far less if nature had not „helped“ science so directly with the help of Alzheimer’s associated mutations. This also shows how enormously important human genetics is to understanding human disease. Even though very few Alzheimer’s cases are caused by genetically inherited mutations, they help us enormously to understand the countless sporadic Alzheimer’s cases. Presenilin would probably still not have been found today if the findings of human genetics had not been available to us. The importance of understanding g-secretase for future therapies will now be discussed below.
Implications for Alzheimer’s disease therapy
Can this knowledge now be exploited to prevent the onset of Alzheimer’s disease?
The clear goal of any Alzheimer’s therapy should be to combat the actual cause. This is certainly, if we start from the amyloid cascade discussed above, to be sought in the aggregation of the amyloid. A reduction of amyloid formation should stabilize the memory performance of patients. This is supported by extensive preclinical evidence demonstrating a direct correlation between the loss of cognitive performance and the volume of aggregated amyloid. Furthermore, the formation of toxic tangles induced by amyloid aggregates is consistently observed within these in vivo systems. At the same time, the formation of toxic tangles induced by amyloid aggregates is consistently observed within these in vivo systems.
If, for example, genetic tricks are used to remove the blades of g-secretase (the presenilins) or screw and nut (nicastrin/pen-2), amyloid production and thus plaque formation are actually completely inhibited. At the same time, cognitive performance within these models is stabilized. Thus, g-secretase represents a key therapeutic target for the development of future drugs. Of course, individual g-secretase genes cannot simply be switched off in humans by genetic manipulation or gene therapy. Therefore, small chemical substances have begun to be developed that insert themselves between the blades of the g-secretase and thus prevent it from snapping shut. This principle follows the very successful development of similar „scissor blockers“ as used to fight AIDS. Of course, g-secretase must not be completely blocked, since it also has a biological function. In fact, g-secretase has a very important biological function. In fact, $\gamma$-secretase has a very important biological function. If individual subunits (blade, screw, or nut) of $\gamma$-secretase are removed in vivo, severe developmental disorders and embryonic lethality will result. However, g-secretase is also needed in the adult organism. Similar to embryonic development, stem cells have the ability to differentiate into functionally distinct „finished“ cells.
This applies, for example, to a variety of different blood cell types. This process is regulated by g-secretase during both embryonic development and within the adult organism. It is therefore not surprising that initial trials in humans with such g-secretase inhibitors resulted in considerable side effects and that these studies had to be discontinued for safety reasons. Does this now take away our hope for rapid drug development? Certainly, this is quite a bitter setback. However, side effects are to be expected in the development of any drug, and any sleeping pill has a fatal effect in too high a dose.
So the next step is to find the right therapeutic window at which amyloid production is sufficiently slowed down, but biological function is still preserved at least to a minimum. In principle, this is achievable, as initial preclinical studies have demonstrated. Furthermore, a number of novel g-secretase inhibitors are currently under development, with the aim of preserving the enzyme’s physiological function. These include, for instance, anti-inflammatory agents such as ibuprofen. Additionally, inhibitors targeting the second secretase, ß-secretase, are being developed (Fig. 3). Expectations here are high, as in vivo models have shown no apparent side effects to date, even following complete blockade. Unfortunately, however, in this case it seems to be technically very difficult for chemists to assemble the appropriate inhibitors in the test tube. Thus, it will certainly be some time before the first safe secretase inhibitors are available. Nevertheless, research to date must be regarded as one of the greatest successes of modern biomedicine. We finally succeeded in identifying all important targets and cloning the corresponding genes. We now have all the necessary targets and model systems in hand to develop effective and safe drugs – a development that all Alzheimer’s researchers hardly dared to dream of 10-15 years ago.
A vaccination against Alzheimer’s?
At the end, I would now like to discuss another spectacular therapeutic approach. In this approach, Alzheimer’s model systems are immunized with the disease-causing substance, amyloid-β. This triggers the production of antibodies against the amyloid in vivo. These reach the brain via yet-to-be-defined pathways, where they interact with amyloid-β plaques and their precursors. Antibody-labeled plaques are then recognized by specific immune cells, which effectively clear the deposits. If this immunization occurs before the initial plaques develop, their formation is entirely prevented—a promising prophylactic strategy against Alzheimer’s disease!
First experiments in humans led at least in a very small study to the still preliminary (and to be interpreted with caution!) result: All patients (only severe Alzheimer cases were included in the study), who responded to the vaccination with a strong production of anti-amyloid antibodies, showed a stabilization of their memory performance in the first two years after the vaccination. Furthermore, it was found that in the brains of these patients, very few plaques were apparently present in the regions otherwise affected by Alzheimer’s disease. This is a highly spectacular result, which naturally gives rise to great hope. But here, too, caution is called for, because unfortunately side effects occurred – about 7% of the vaccinated patients developed severe brain inflammation, which led to the immediate termination of the study. It should be emphasized, however, that contrary to what has been reported in the press, not a single patient died as a result of immunization.
As with g-secretase inhibitors, alternative methods of immunization are now being tried which it is hoped will not cause inflammation. Attempts are being made to circumvent the side effects with so-called passive vaccination, in which industrially produced antibodies are injected into the bloodstream. The outcomes of in vivo studies were sufficiently encouraging to justify the next step into clinical application. The first results of this landmark study were anticipated as early as 2006.
Is there hope?
I think that is the all-important question. If we do not manage to get Alzheimer’s disease under control in time, we will be faced with the probably insoluble task of caring for millions of dementia patients. Basic research from numerous different disciplines such as cell biology, molecular biology, pharmacy, biochemistry and physics have managed to clarify the mechanisms of action of Alzheimer’s disease, at least partially, within a very short time in huge interdisciplinary national and worldwide efforts. However, much is still unclear, such as the toxic mechanisms of action of amyloid, but with the identification of numerous targets and the first pioneering therapeutic approaches, we are, despite all the problems, on the right path to getting one of the biggest health problems under control.
Es bleibt aber zu hoffen, dass in Deutschland endlich die Forschung an der Alterung und allen mit ihr verbundenen Problemen zielgerichtet gefördert wird. Ein nationales interdisziplinäres Forschungszentrum ist hierzu unbedingt notwendig.
Abbildungen
Abb. 1:
Die Amyloidplaquepathologie.
Weite Bereiche des Gehirns sind mit Amyloidplaques (kleine braune Flecken) übersäht (linkes Bild). Bei größerer Vergrößerung (rechtes Bild) sieht man schwarz gefärbte absterbende oder bereits tote Nervenzellen um den zentralen Plaque angeordnet. Die schwarzen Strukturen werden Tangles genannt. Tangles werden durch Amyloidplaques bzw. deren Vorstufen induziert.
Abb. 2:
Sekretasen sind molekulare Scheren
Die g-Sekretase ist wie eine Papierschere aus zwei Klingen (den Presenilinen), einer Schraube (Nicastrin) und einer Mutter (Pen-2) aufgebaut.
Abb. 3:
Das Amyloid wird durch die scherenartigen Sekretasen aus einem Vorläufer herausgeschnitten.
By Professor Christoph Hock, MD, Co-Director and Chief Physician of the Department of Psychiatric Research at the University of Zurich.
About 8% of people over 65 suffer from gradual memory decline. Nerve cells are no longer functional and die, and the exchange of information between brain cells fails. This is triggered by protein deposits in the brain, so-called beta-amyloid plaques. It is not yet known exactly how these pathological changes develop. In the meantime, around 20 million people worldwide suffer from Alzheimer’s disease, and the trend is increasing in view of high life expectancy. Although there are drugs that influence the messenger substances that are disturbed in dementia, they only delay the disease for about a year.
Beta-amyloid as a pharmacological target of a vaccination?
The human immune system is capable of fighting and eliminating bacteria, viruses, parasites and toxic pollutants. Of central importance is the distinction between endogenous and exogenous. The beta-amyloid plaques of Alzheimer’s disease contain beta-amyloid peptides, derivatives of the endogenous beta-amyloid precursor protein, APP, as their main component. Vaccination against beta-amyloid therefore initially seems counterproductive. However, the aggregation process, which eventually leads to mature beta-amyloid plaques via soluble beta-amyloid peptides and various intermediates, such as oligomers and protofibrils, is associated with the formation of new, fibrillar, nonphysiological structures. This leads to the presentation of neo-epitopes that are distinct from physiological epitopes of beta-amyloid peptides and thus may be suitable targets for immunization.
Passive and active immunotherapy
Immunization against an antigen can take active and passive forms. The basic principle of active immunization is based on the presentation of beta-amyloid or derivatives as antigen in combination with an immunogenic adjuvant, together the „vaccine“. After injection of the vaccine, recognition of the antigen as foreign to the body occurs with subsequent humoral (formation of antibodies by B lymphocytes) and cellular (formation of antigen-specific T cells) immune responses. In passive immunization, biotechnologically produced, usually „humanized“, monoclonal antibodies against beta-amyloid are administered directly by injection, e.g. into venous blood („intravenously“) or into subcutaneous fatty tissue („subcutaneously“).
Therapy research in model systems
Experimental models are often used to research new therapeutic approaches to human diseases, ranging from in vitro systems to complex in vivo models. In a transgenic preclinical model of Alzheimer’s disease, immunization therapy involving multiple injections against amyloid-β was first attempted in 1999, with surprising success: the vaccination triggered a robust immune response within the model system, leading to the formation of antibodies—the body’s own defense mechanisms—against beta-amyloid. As a result, a reduction of beta-amyloid plaques and a remission of learning disorders could be measured. It was shown that the formation of antibodies after vaccination was the decisive step towards therapeutic success.
The first clinical trials: problematic side effects
However, in the first clinical trials in patients with this approach, significant side effects in the form of aseptic brain inflammation occurred in 6% of the study patients, so that the vaccinations had to be discontinued. Clinical and neuropathological follow-up examinations of the immunized patients confirmed on the one hand the inflammatory side effects, probably an expression of an excessive cellular immune response. On the other hand, however, there were important indications of the specific formation of antibodies against beta-amyloid, the crossing of such antibodies across the blood-brain barrier, the removal of amyloid plaques from the brain, and an improvement in memory performance as well as clinical stabilization in patients who had formed antibodies.
Further development of immunotherapy
Currently, research efforts are focused on more precisely defining the target of antibodies, targeting structural neoepitopes, and better ruling out autoimmune reactions. Preclinical trial designs are being developed to minimize the risk of inflammatory reactions, autoimmune diseases and bleeding. There is high activity of clinical trial programs, including predominantly passive immunization approaches, but also active vaccination. The development stages of clinical programs range (as of this writing, October 2008) from Phase I (e.g., CAD106, Novartis; R1450, Roche; PF04360365, Pfizer; V950, Merck; GSK933776A, GlaxoSmithKline), to Phase II (e.g., LY2062430, Eli Lilly; IVIg-mix/Gammaguard, Baxter/Cornell), to Phase III (Bapineuzumab, Elan/Wyeth). The goal of the programs is to demonstrate significant clinical efficacy while maintaining adequate safety and tolerability of immunotherapy.
As part of the German Foundation Day 2016 in Leipzig, Roland Bergfeld, Chairman of the Board of the Hans and Ilse Breuer Foundation, took part in an expert discussion on age-related brain diseases. He as well as Prof. Dr. med. Thomas Gasser, Chairman of the Board of the Hertie Institute for Brain Research and Director of Neurology at the University of Tübingen, Monika Kaus, Chairwoman of the Board of the German Alzheimer Society and Prof. Dr. Dr. h. c. Andreas Kruse, Director of the Institute for Gerontology at the University of Heidelberg, provided insights into challenges and perspectives from their respective contexts in the panel moderated by Julia Riedel (Hertie Foundation).
It became clear that our society is on the right track in many areas, but that a great deal remains to be done and how closely the requirements are interrelated.
By loading this video, you agree to the privacy policy of Youtube.