Von Prof. Dr. Christian Haass, Labor für Alzheimer- und Parkinsonforschung, Lehrstuhl für Stoffwechsel-Biochemie an der Ludwig-Maximilians-Universität, München
Vor fast genau 100 Jahren kam in die Praxis des Münchner Psychiaters Dr. Alois Alzheimer eine Patientin, die ihre Erkrankung mit folgenden erschütternden Worten beschrieb: „Ich habe mich selbst verloren“. Diese Patientin sollte der weltweit erste Alzheimer-Fall sein und ist heute bekannt als der berühmte Fall der „Auguste D.“ Natürlich gab es schon damals viele andere Fälle der Altersvergesslichkeit, wie sie auch sehr treffend in Paul Klees „Der vergessliche Engel“ künstlerisch dargestellt ist. Heute ist die Alzheimer- Erkrankung weltweit die häufigste Demenz. Allein in Deutschland sind zurzeit fast 1,2 Millionen Alzheimer-Patienten bekannt. Da unser Durchschnittsalter wegen des enormen medizinischen Fortschrittes massiv zugenommen hat (und auch noch weiter steigt), müssen wir mit einer wahren Flut von Alzheimerfällen rechnen.
Altersheime, Pflegeanstalten und Krankenkassen sind auf dieses Problem nicht vorbereitet. Die Entwicklung einer wirkungsvollen Therapie gegen diese „Volkserkrankung“ ist unbedingt notwendig. Die derzeitigen Medikamente können die Symptome nur vorübergehend ein wenig mildern, den schleichenden Gedächtnisschwund aufhalten können sie aber keineswegs. Um das zu erreichen, ist ein genaues Verständnis der molekularen und zellbiologischen Wirkungsmechanismen notwendig, so genannte target molecules müssen identifiziert werden, deren Blockierung (Inhibierung) den Verlauf der Erkrankung verlangsamt oder gar vollständig stoppt.
Um dies zu erreichen, wollen wir verstehen, wie es zu der pathologischen Ablagerung der kennzeichnenden Amyloid-Plaques im Alzheimer-Gehirn kommt. Unsere Hypothese ist, dass eine Blockade der Plaquebildung das Fortschreiten der Erkrankung unterbinden sollte und vielleicht sogar, bei rechtzeitigem Einschreiten, von vornherein verhindert. Die Amyloid- Plaques bestehen aus einem kleinen Eiweiß, dem Amyloid. Das Amyloid selbst wird offenbar aus einem sehr viel größeren Eiweiß herausgeschnitten, woraufhin es verklumpt, kleine Aggregate bildet und sich schließlich in den Plaques ansammelt. Die kleinen Aggregate und die Plaques selbst schädigen umliegende Nervenzellen so schwerwiegend, dass diese letztendlich absterben. Der über die Jahre hinweg steigende Verlust von Nervenzellen führt dann zu dem dramatischen Gedächtnisverlust der Patienten.
Noch während meiner Zeit als Assistant Professor an der Harvard-Universität in Boston machte ich bei der Untersuchung einfacher menschlicher Zellen die sehr überraschende Entdeckung, dass die Entstehung des Amyloids ein völlig normaler Vorgang ist. Denn er läuft bei uns allen vom Tag der Geburt an ständig ab. Leider tickt damit aber in uns eine Zeitbombe, die im hohen Alter, wenn genügend Amyloid akkumuliert ist, zu einer explosionsartigen Zunahme der Plaques und des Nervenzelltodes führt. Diese Tatsache lehrt, dass uns die Erkrankung in der Tat alle treffen kann und treffen wird – wenn wir nur alt genug werden. Im Labor benutzen wir diese Entdeckung, um die genauen Mechanismen des Herausschneidens von Amyloid zu verstehen. Hierzu arbeiten wir mit menschlichen Zellen, der Hefe, dem Zebrafisch und genmanipulierten (transgenen) Mäusen.
Unser Ziel war es, die „scherenartigen“ Enzyme zu finden, welche das giftige Amyloid herausschneiden. Diese Scheren bezeichnen wir heute als „Sekretasen“. Es gibt für jeden der zwei Schnitte je eine typische Sekretase (die β-, und die γ-Sekretase), die wir detailliert auf ihre Funktionsweise untersucht haben. Wir fanden mit Hilfe der simplen Bäckerhefe heraus, dass die γ-Sekretase völlig überraschend aufgebaut ist. Sie ähnelt in ihrer Struktur, Bau- und Funktionsweise tatsächlich einer herkömmlichen Papierschere. Wir fanden zwei Schneiden, die sich auf je zwei verschiedenen Untereinheiten befinden und durch eine Schraube und eine Mutter zusammengehalten werden. Mit Hilfe einer modernen gentechnologischen Methode gelang es uns, gezielt die Schraube herauszudrehen. Dies hatte zur Folge, dass die beiden Schneiden auseinanderfielen und von der Zelle als Abfall erkannt und abgebaut wurden. Das Fehlen der aktiven γ-Sekretase hatte dann ganz massive Folgen für die Entstehung des giftigen Amyloids. Seine Produktion war fast vollständig blockiert, d.h., wir hatten tatsächlich das richtige Zielmolekül identifiziert und eine Grundlage bereitet zur gezielten Entwicklung von Medikamenten.
Wir suchen nun mit Hilfe voll automatisierter Tests kleine chemische Verbindungen, welche das Zuschnappen der Scheren gezielt und selektiv verhindern. Gleichzeitig haben wir natürlich ein großes Interesse daran, herauszufinden, welche normalen Funktionen die Scheren in unserem Körper haben. Dies ist nicht nur von enormem akademischem Interesse, sondern spielt auch bei der Therapie eine höchst bedeutende Rolle, da wir ja keinesfalls essenzielle Funktionen im menschlichen Körper blockieren dürfen. Die biologische Funktion der Scheren haben wir in einfachen Modellsystemen untersucht. Hierzu haben wir kürzlich die Zebrafischtechnologie mit Hilfe des Leibnizpreises der DFG und der tatkräftigen Unterstützung der LMU eingeführt. Dieses System erlaubte es uns, nicht nur die normale Funktion der Schere zu identifizieren (in der Zelldifferenzierung), sondern auch Nebenwirkungen von Hemmstoffen – so genannten Inhibitoren – der Scheren zu beschreiben. Unser Modell eignet sich daher nun ganz hervorragend, um gezielt solche Medikamente zu finden, welche keine der unerwünschten Nebenwirkungen zeigen. Weiterhin sind wir dabei, den Fisch genetisch so zu manipulieren, dass er eine Alzheimer-Symptomatik inklusive Gedächtnisverlust zeigt (ein Fisch erinnert sich z. B. durchaus daran, in welcher der vier Ecken eines Aquariums er sein Futter bekommt). Das Genom dieses Fisches werden wir dann so verändern, dass jedes Gen einzeln verändert wird. Um das zu ermöglichen, haben wir ein großes Fischhaus eingerichtet, in dem im Moment über 1000 Aquarien stehen.
Ziel ist es, künstliche Mutanten zu finden, bei denen die Alzheimer-Symptomatik entweder abgeschwächt ist oder verstärkt auftritt. Von der Klonierung der entsprechenden Gene versprechen wir uns, neue Einblicke in bisher noch völlig unbekannte Aspekte der Alzheimer-Erkrankung zu erlangen, in die Mechanismen, die zum Tode der Nervenzellen durch Amyloidaggregation führen.
Unsere Arbeiten führen wir in lokalen (SFB 596 Molekulare Mechanismen der Alzheimer- Erkrankung), nationalen (DFG, Schwerpunkt Zellbiologie der Alzheimer-Erkrankung), europäischen (European Community Grants) und amerikanischen (American Health Assistance Foundation) Verbundsystemen durch. Es bestehen aktive Kooperationen mit Pharmafirmen (Boehringer Ingelheim KG; Merck Sharp & Dome; Hofmann La Roche).
Von Prof. Dr. Christian Haass, Labor für Alzheimer- und Parkinsonforschung, Lehrstuhl für Stoffwechselbiochemie an der Ludwig-Maximilians-Universität, München
Die Alzheimer Erkrankung ist weltweit die häufigste Form von Demenz, und nahezu jeder Leser dieses Artikels kennt im Kreise seiner Freunde und Verwandten einen Menschen der von dieser Geißel des 20. und 21. Jahrhunderts befallen ist. Das immer häufigere Auftreten der Alzheimer Erkrankung hängt mit der dramatisch steigenden Lebenserwartung zusammen. Je älter wir werden, umso höher ist leider die Chance erste Symptome zu entwickeln. Was sich anfänglich durch eine geringfügige Vergesslichkeit auszeichnet, kann im Endstadium bis zum vollständigen Verlust der Persönlichkeit führen – ein unvorstellbares Drama für Betroffene und deren Angehörige. Auf Grund der altersabhängigen Zunahme der Alzheimer Erkrankung, werden wir bei zunehmend höherem medizinischen Standards mit einer massiven Explosion der Patientenzahlen rechnen müssen, eine Tatsache auf die Gesundheitspolitiker in Deutschland noch nicht vorbereitet sind. Es fehlt an Personal, Pflegeheimen und finanziellen Mitteln für die Pflege dementer Patienten. Interdisziplinäre Forschungszentren, die sich gezielt mit den Mechanismen altersbezogener Erkrankungen beschäftigen gibt es in Deutschland nicht.
Den ersten nationalen Verbund deutscher Alzheimerforscher konnte ich erst vor fünf Jahren mit Hilfe der Deutschen Forschungsgemeinschaft (DFG) zusammenstellen, eine Initiative, die in den USA in den 80er Jahren bereits gestartet wurde. Dies ist umso erstaunlicher, da einige der grundlegenden und aufregendsten Entdeckungen zu den Mechanismen der Alzheimer Erkrankung in Deutschland erfolgten. Ich erwähne hier nur die ursprüngliche Entdeckung der Pathologie durch Alois Alzheimer (vor 100 Jahren), die Klonierung des „Alzheimergens“ und die Identifizierung eines der zentralen Alzheimerenzyme auf das ich später noch genauer eingehen werde.
Im Folgenden werde ich erst die Pathologie der Erkrankung schildern und dann die dafür verantwortlichen molekularen Mechanismen darstellen um dann am Ende auf neue Möglichkeiten der Therapie und den damit zusammenhängenden Hoffnungen und Schwierigkeiten einzugehen.
Aggregierende Amyloide lösen eine tödliche Kaskade aus
Pathologisch ist die Alzheimer Erkrankung unter anderem durch das Auftreten zahlloser Amyloid Plaques im Gehirn der Patienten gekennzeichnet (Abb. 1). In manchen Regionen des Gehirns können diese Plaques bis zu 20% des Gehirnvolumens einnehmen. Während sich die Plaques außerhalb der Nervenzellen befinden, findet man in zahlreichen Nervenzellen weitere Verklumpungen, die wir Tangles nennen (Abb. 1). In den betroffenen Regionen des Gehirns kommt es zu einem dramatischen Verlust von Nervenzellen. Amyloid Plaques enthalten also offenbar ein Molekül, das umliegende Nervenzellen tötet. Dieses Molekül entpuppte sich als ein kleines Eiweiß, das wir heute Amyloid ß-Peptid (Amyloid) nennen. Amyloid neigt dazu mit sich selbst zu aggregieren und dann als unlöslicher Komplabgelagert zu werden. Solche aggregierende zytotoxische Peptide sind ein typisches Merkmal vieler neurodegenerativer Erkrankungen, wobei bei jeder Erkrankung ein spezifisches Amyloid Molekül gebildet wird. Die Amyloide der unterschiedlichsten Erkrankungen scheinen jedoch eines gemeinsam zu haben: durch eine Umfaltung ihrer natürlichen dreidimensionalen Struktur wird die Aggregation und die daraus folgende Präzipitation gefördert. Die Akkumulation dieser Aggregate bestehend aus den unterschiedlichsten Amyloiden scheint dann den neuronalen Zelltod einzuleiten.
Im Falle der Alzheimer Erkrankung lösen kleine oligomere Amyloid Aggregate (Vorstufen zu den eigentlichen Plaques) eine tödliche Kaskade aus, die heute unter dem Namen Amyloidkaskade bekannt ist. Hierbei wird unter u.a. die Tangle Bildung hervorgerufen (schwarze „Bündel“ in Abb.1 rechts), die wiederum die Bildung zahlreicher giftiger Moleküle induzieren, deren Folge dann das Absterben von Nervenzellen ist. Darüber hinaus haben die kleinen Amyloidoligomere auch eine direkte Wirkung auf unsere Gedächtnisvorgänge. Breits in geringsten Spuren können Sie die Manifestation von erlerntem Wissen direkt beeinträchtigen. Da das Amyloid nach unseren heutigen Kenntnissen die Amyloidkaskade auslöst ist dieses Molekül ein idealer Angriffspunkt für neue, Ursachen orientierte Therapieformen. Man sucht hierbei nach den Genen und den von ihnen kodierten Enzymen (sog. Targets oder Zielmoleküle), die für die Bildung des Amyloid Moleküls verantwortlich sind. Dieser Ansatz schließt natürlich andere Moleküle, die sich weiter unten in der Kaskade befinden (wie z. B. die Tangles) nicht aus.
Die familiäre Alzheimer Erkrankung
In einigen wenigen Fällen (maximal 1-5 %) tritt die Alzheimer Erkrankung in einer besonders aggressiven Form innerhalb einzelner Familien auf. Diese Krankheitsvariante ist pathologisch nicht zu unterscheiden von der großen Vielzahl der sporadischen Alzheimer Fälle, sie bricht allerdings sehr früh aus. Es sind Fälle bekannt in denen es im Alter von unter 30 Jahren zu einem vehementen Ausbruch der Alzheimer Erkrankung kam. Eine Häufung von schweren Alzheimer Fällen mit frühem Beginn in einzelnen Familien impliziert daher bereits, daß hier genetische Risikofaktoren vorliegen, die weitervererbt werden. Diese Risikofaktoren scheinen, wenn sie durch Mutationen entsprechend verändert sind, den Ablauf der Alzheimer Pathologie und speziell die Amyloidplaque Bildung dramatisch zu beschleunigen. Für Molekularbiologen enthalten diese Gene daher den Schlüssel zum Verständnis der zellulären Mechanismen der Erkrankung, obwohl natürlich die Anzahl der genetisch vererbten Alzheimerfälle epidemiologisch irrelevant ist.
Im Folgenden sollen nun Alzheimer verursachende Mutationen in drei Genen beschrieben werden, und am Ende sollen jüngste Forschungsergebnisse dargestelltwerden, die zeigen daß die Inaktivierung von bestimmten Alzheimergenen zu einer massiven Reduktion der Amyloid Bildung führen. Mutationen in drei Genen beschleunigen die Aggregation des Amyloid Moleküls.
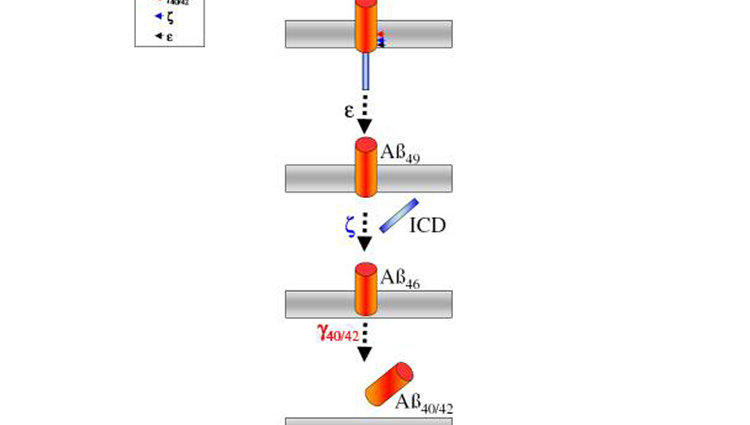
Die ersten Mutationen wurden im Gen des Amyloid Vorläufers selbst befunden. Diese Mutationen liegen nicht etwa verstreut über das gesamte ßAPP Molekül, sondern treten exakt an den Schnittstellen der drei Sekretasen auf. Letztendlich bewirken alle diese Mutationen ein und dasselbe: sie verursachen die Produktion eines minimal veränderten Amyloid Moleküls. Im Gegensatz zu dem 40 Bausteine (Aminosäuren) langen Aß, das wir alle ständig produzieren, wird durch die Mutationen die Bildung eines um nur zwei Bausteine verlängerten Aß Moleküls verstärkt (Abb. 4). Diese Variante hat die fatale Eigenschaft extrem rasch mit sich selbst zu aggregieren (Abb. 4). Die Mutationen reduzieren daher die Zeit, die normalerweise für die Amyloidplaque Bildung benötigt wird, auf ein Minimum und führen damit zum sehr frühen Ausbruch der Erkrankung, ohne jedoch die charakteristische Pathologie als solches zu verändern.
Wesentlich häufigere Mutationen wurden in den beiden sehr ähnlichen Presenilingenen, (abgekürzt PS1 und PS2) gefunden. Alle bisher untersuchten Mutationen verursachen, genau wie die Mutationen im Amyloid Vorläufer, eine verstärkte Bildung des um zwei Aminosäuren verlängerten Amyloid Moleküls (Abb. 4). Damit kommt es auch hier wieder zur beschleunigten Bildung der Alzheimerpathologie, die dann bereits in der zweiten Lebensdekade auftreten kann. In der Natur treten damit Veränderungen in Genen auf, die ganz offensichtlich (wie im Falle des Vorläufer Gens) die Amyloid Bildung direkt beeinflussen. Die viel häufigeren Mutationen in den Presenilinen waren aber sehr schwer zu erklären. Was haben die Preseniline mit der Amyloid Bildung zu tun? Wie beeinflussen dies die Entstehung des Amyloides?
Wesentlich häufigere Mutationen wurden in den beiden sehr ähnlichen Presenilingenen, (abgekürzt PS1 und PS2) gefunden. Alle bisher untersuchten Mutationen verursachen, genau wie die Mutationen im Amyloid Vorläufer, eine verstärkte Bildung des um zwei Aminosäuren verlängerten Amyloid Moleküls (Abb. 4). Damit kommt es auch hier wieder zur beschleunigten Bildung der Alzheimerpathologie, die dann bereits in der zweiten Lebensdekade auftreten kann. In der Natur treten damit Veränderungen in Genen auf, die ganz offensichtlich (wie im Falle des Vorläufer Gens) die Amyloid Bildung direkt beeinflussen. Die viel Die Preseniline sind ein wichtiger Baustein der g-Sekretase.
Die g-Sekretase wurde als Schlüsselenzym der Alzheimererkrankung über Jahrzehnte hinweg von unzähligen Labors in zum Teil sehr aufwändigen (aber dennoch erfolglosen) Experimenten gesucht. Wie immer lag der Schlüssel in einer denkbar einfachen Schlussfolgerung. Schauen wir uns noch einmal den Effekt den Effekt der Mutationen in den Presenilin Genen auf die Amyloidbildung an (Abb. 4). Weit über 100 verschiedene Mutationen verteilt über das gesamte Gen haben alle merkwürdiger Weise ein und denselben Effekt: Sie verschieben den Schnitt der g-Sekretase um exakt 2 Bausteine, so dass ein längeres, schneller aggregierendes Amyloid entsteht. Wie können nun diese Mutationen die g-Sekretase so tödlich beeinflussen? Die Lösung war denkbar einfach: Die Preseniline sind identisch mit der g-Sekretase, oder zumindest ein wichtiger Baustein der g-Sekretase!
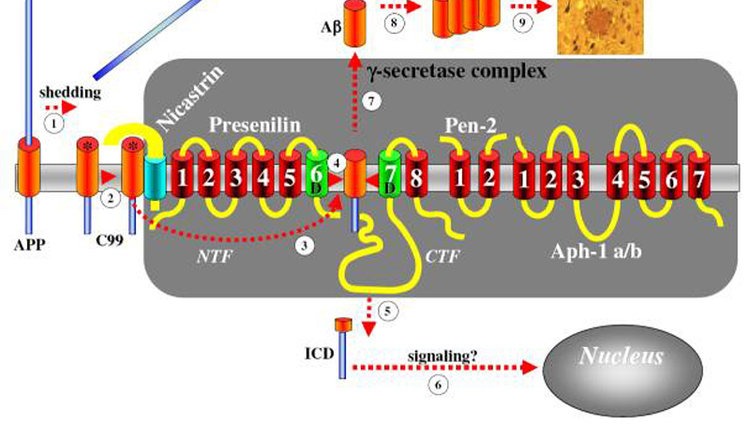
Um den Aufbau der g-Sekretase zu verstehen, sollten wir das Bild einer typischen Papierschere vor Augen haben (Abb. 2). Diese besteht aus zwei Klingen, die den eigentlichen Schneidevorgang durchführen. Die Klingen sind identisch mit den Presenilinen. Diese liegen immer in Form von zwei getrennten Teilen vor, von denen beide für den Schnittvorgang absolut notwendig sind. Die voneinander getrennten Klingen müssen aber auch zusammen gehalten werden. Hierfür werden in der Mechanik Schrauben und Muttern verwendet (Abb. 2). Genau das hat die Evolution „kopiert“. Zwei weitere Gene (bzw. deren Proteinprodukte) werden nämlich benötigt um die Schere aufzubauen und zusammen zu halten. Die Schraube wird dabei durch ein Protein namens Nicastrin und dzu halten. Die Schraube wird dabei durch ein Protein namens Nicastrin und die Mutter durch Pen-2 gebildet (Abb. 2). Beide zusammen halten die Klingen der Sekretase zusammen und erlauben deren präzises Zuschnappen.
Unser heutiges Verständnis der g-Sekretase (und damit der Amyloid Bildung generell) wäre weit geringer, wenn die Natur nicht mit Hilfe der Alzheimer assoziierten Mutationen der Wissenschaft so direkt „geholfen“ hätte. Dies zeigt auch wie enorm wichtig die Humangenetik für das Verständnis menschlicher Erkrankung ist. Auch wenn nur sehr wenige Alzheimer Fälle durch genetisch vererbte Mutationen hervorgerufen werden, helfen uns diese jedoch enorm auch die unzählig vielen sporadischen Alzheimer Fälle zu verstehen. Presenilin wäre vermutlich heute noch immer nicht gefunden, wenn uns nicht die Erkenntnisse der Humangenetik zur Verfügung gestanden hätten. Wie wichtig das Verständnis der g-Sekretase für zukünftige Therapien wird im Folgenden nun diskutiert werden.
Implikationen für eine Therapie der Alzheimer Erkrankung
Kann man dieses Wissen nun ausnutzen um den Ausbruch der Alzheimer Erkrankung zu verhindern?
Klares Ziel einer jeden Alzheimertherapie sollte es sein, die eigentliche Ursache zu bekämpfen. Diese ist sicherlich, wenn wir von der oben besprochenen Amyloidkaskade ausgehen, in der Aggregation des Amyloids zu suchen. Eine Reduktion der Amyloidbildung sollte die Gedächtnisleistung von Patienten stabilisieren. Diese könnte in der Tat der Fall sein, denn eine Vielzahl von Versuchen in Tiermodellen belegen einen direkten Zusammenhang zwischen dem Verlust der Gedächtnisleistung und der Menge an aggregiertem Amyloid. Gleichzeitig wird die Bildung der giftigen Tangles durch Amyloid Aggregate im Tiermodell regelrecht induziert.
Entfernt man nun z. B. mit Hilfe genetischer Tricks die Klingen der g-Sekretase (die Preseniline) oder Schraube und Mutter (Nicastrin/Pen-2) wird tatsächlich die Amyloid Produktion und damit die Plaquebildung vollständig inhibiert. Gleichzeitig werden die Gedächtnisleistungen der Tiere stabilisiert. Damit ist die g-Sekretase mit Sicherheit ein wichtiges „Target“ (Zielmolekül) für die Entwicklung zukünftiger Medikamente. Natürlich lassen sich im Menschen einzelne Gene der g-Sekretase nicht einfach durch genetische Manipulation oder Gentherapie ausschalten. Man hat daher begonnen kleine chemische Substanzen zu entwickeln, die sich zwischen die Klingen der g-Sekretase einlagern und diese so am Zuschnappen hindern. Dieses Prinzip folgt der sehr erfolgreichen Entwicklung ähnlicher „Scherenblockierer“ wie bei der AIDS Bekämpfung. Natürlich darf man die g-Sekretase nicht vollständig blockieren, da diese ja auch eine biologische Funktion hat. Die g-Sekretase hat in der Tat eine sehr wichtige biologische Funktion. Entfernt man in Mäusen oder anderen Modellorganismen einzelne Untereinheiten (Klinge, Schraube oder Mutter) der g-Sekretase kommt es zu schweren Entwicklungsstörungen und zum Absterben des Embryos. Aber auch im adulten Organismus wird die g-Sekretase gebraucht. Ähnlich wie bei der Embryonalentwicklung, haben auch hier Stammzellen die Möglichkeit in funktionell unterschiedliche „fertige“ Zellen zu differenzieren.
Dies betrifft z. B. eine Reihe verschiedener Blutzellen. Dieser Vorgang wird nun bei der Embryonalentwicklung wie auch bei adulten Tieren durch die g-Sekretase gesteuert und reguliert. Es ist daher nicht verwunderlich, dass es bei ersten Versuchen am Menschen mit solchen g-Sekretaseinhibitoren zu erheblichen Nebenwirkungen kam und diese Studien aus Sicherheitsgründen abgebrochen werden musste. Nimmt uns das nun die Hoffnung auf eine rasche Medikamententwicklung? Sicherlich ist das ein recht herber Rückschlag. Es ist aber bei der Entwicklung von jedem Medikament mit Nebenwirkungen zu rechnen, jedes Schlafmittel hat in zu hoher Dosis eine fatale Wirkung.
Man muss also im nächsten Schritt das richtige therapeutische Fenster finden, bei dem die Amyloidproduktion genügend gebremst wird, aber die biologische Funktion auch noch zumindest in einem Mindestmaß erhalten bleibt. Prinzipiell ist das möglich, wie erste Versuche an Tiermodellen gezeigt haben. Darüber hinaus werden zurzeit eine Reihe neuartiger g-Sekretaseinhibitoren entwickelt, von denen man sich erhofft, dass sie die biologisch normale Funktion der g-Sekretase nicht reduzieren. Hierzu gehören z. B. die Entzündung hemmenden Mittel wie Ibuprofen. Weiterhin werden momentan auch gegen die zweite Sekretase, die ß-Sekretase (Abb. 3) Inhibitoren entwickelt. Hier haben wir große Hoffnungen, da bisher im Tiermodell, auch nach vollständiger Blockade keinerlei offensichtliche Nebenwirkungen auftraten. Leider scheint es aber in diesem Fall für die Chemiker technisch sehr schwierig zu sein die passenden Inhibitoren im Reagenzglas zusammenzubauen. Damit wird es sicherlich noch etwas dauern, bis die ersten sicheren Sekretase Inhibitoren zur Verfügung stehen. Dennoch muss man die bisherige Forschung als einen der größten Erfolge der modernen Biomedizin betrachten. Es gelang letztendlich alle wichtigen Targets zu identifizieren und die entsprechenden Gene zu klonieren. Wir haben jetzt alle notwendigen Zielmoleküle und Modellsysteme in der Hand um wirksame und sichere Medikamente zu entwickeln – eine Entwicklung von der alle Alzheimerforscher noch vor 10-15 Jahren kaum zu träumen wagten.
Eine Impfung gegen Alzheimer?
Am Ende möchte ich nun noch einen weiteren spektakulären Therapieansatz diskutieren. Hier impft man Alzheimermäuse mit der krankheitserregenden Substanz, dem Amyloid. Die Maus produziert daraufhin Antikörper gegen das Amyloid. Diese gelangen auf ungeklärten Weg ins Gehirn, wo sie mit dem Amyloidplaques und deren Vorstufen interagieren. Antikörper markierte Plaques werden dann von bestimmten Immunzellen erkennt, welche die Plaques regelrecht auffressen. Impft man die Mäuse noch vor der Entstehung der ersten Plaques, kommt es erst gar nicht zur Plaqueentwicklung – eine prophylaktische Alzheimertherapie!
Erste Experimente im Menschen führten zumindest in einer sehr kleinen Studie zu dem noch vorläufigen (und mit Vorsicht zu interpretierenden!) Ergebnis: Alle Patienten (in der Studie waren nur schwere Alzheimerfälle eingeschlossen), die mit einer starken Produktion von anti-Amyloid Antikörper auf die Impfung antworteten, zeigten in den ersten beiden Jahren nach der Impfung eine Stabilisierung ihrer Gedächtnisleistung. Weiterhin fand man, dass im Gehirn dieser Patienten ganz offenbar in den sonst vom Alzheimer befallenen Regionen nur noch sehr wenige Plaques zu finden waren. Das ist ein höchst spektakuläres Ergebnis, das natürlich Anlass zu größten Hoffnungen gibt. Aber auch hier ist Vorsicht angebracht, denn es traten leider Nebenwirkungen auf – ca. 7% der geimpften Patienten entwickelten schwere Hirnentzündungen, die zu einem sofortigen Abbruch der Studie führten. Es sollte aber betont werden, dass anders als in der Presse beschrieben, kein einziger Patient an den Folgen der Immunisierung gestorben ist.
Wie bei den g-Sekretase Inhibitoren werden nun alternative Methoden der Immunisierung probiert, von denen man sich erhofft, dass sie keine Entzündungen hervorrufen. Man versucht mit der so genannten passiven Impfung, bei der industriell hergestellte Antikörper in die Blutbahn gespritzt werden, die Nebenwirkungen zu umgehen. Tierversuche verliefen so positiv, dass man den erneuten Schritt in den Patienten bereits gewagt hat. Erste Ergebnisse dieser Studie sind in 2006 zu erwarten.
Gibt es Hoffnung?
Ich glaube das ist die alles entscheidende Frage. Schaffen wir es nicht rechtzeitig die Alzheimererkrankung in den Griff zu bekommen, stehen wir vor der wohl unlösbaren Aufgabe der Pflege von Millionen von Demenzpatienten. Die Grundlagenforschung aus zahlreichen verschiedenen Disziplinen wie der Zell Biologie, der Molekularen Biologie, der Pharmazie, der Biochemie und der Physik haben es in riesigen interdiszipinären nationalen und weltweiten Anstrengungen geschafft innerhalb kürzester Zeit die Wirkungsmechanismen der Alzheimer Erkrankung zumindest teilweise aufzuklären. Vieles ist aber noch unklar, wie z. B. die toxischen Wirkungsmechanismen des Amyloid, aber mit der Identifizierung zahlreicher Targets und ersten richtungweisenden Therapieansätzen sind wir trotz aller Probleme auf dem richtigen Weg eines der größten Gesundheitsprobleme in den Griff zu bekommen.
Es bleibt aber zu hoffen, dass in Deutschland endlich die Forschung an der Alterung und allen mit ihr verbundenen Problemen zielgerichtet gefördert wird. Ein nationales interdisziplinäres Forschungszentrum ist hierzu unbedingt notwendig.
Abbildungen
Abb. 1:
Die Amyloidplaquepathologie.
Weite Bereiche des Gehirns sind mit Amyloidplaques (kleine braune Flecken) übersäht (linkes Bild). Bei größerer Vergrößerung (rechtes Bild) sieht man schwarz gefärbte absterbende oder bereits tote Nervenzellen um den zentralen Plaque angeordnet. Die schwarzen Strukturen werden Tangles genannt. Tangles werden durch Amyloidplaques bzw. deren Vorstufen induziert.
Abb. 2:
Sekretasen sind molekulare Scheren
Die g-Sekretase ist wie eine Papierschere aus zwei Klingen (den Presenilinen), einer Schraube (Nicastrin) und einer Mutter (Pen-2) aufgebaut.
Abb. 3:
Das Amyloid wird durch die scherenartigen Sekretasen aus einem Vorläufer herausgeschnitten.
Abb. 4:
Familiär vererbte Mutationen beeinflussen die Präzision des g-Sekretase Schnittes. Der Schnitt wird um zwei Bausteine verschoben (grüner Pfeil), so dass ein längeres Amyloid entsteht, das dann schneller aggregiert.
Hier können Sie sich den Beitrag im PDF-Format herunterladen… (PDF-Dokument, 67.2 KB)
Laden Sie sich hier die Abbildungen 1 und 2 als PDF herunter… (PDF-Dokument, 224.7 KB)
Von Professor Dr. med. Christoph Hock, Co-Direktor und Chefarzt der Abteilung für psychiatrische Forschung an der Universität Zürich
Rund 8% der über 65jährigen leiden unter einem allmählichen Abbau des Gedächtnisses. Nervenzellen sind nicht mehr funktionstüchtig und sterben ab, der Informationsaustausch zwischen Gehirnzellen versagt. Auslöser sind Proteinablagerungen im Hirn, so genannte beta-Amyloid-Plaques. Noch ist nicht genau bekannt, wie diese krankhaften Veränderungen entstehen. Mittlerweile leiden an die 20 Millionen Menschen weltweit an der Alzheimer-Krankheit, die Tendenz steigt angesichts der hohen Lebenserwartung. Zwar gibt es Medikamente, welche die bei Demenz gestörten Botenstoffe beeinflussen, doch sie verzögern die Krankheit lediglich für etwa ein Jahr.
Beta-Amyloid als pharmakologischer Angriffspunkt einer Impfung?
Das Immunsystem des Menschen ist in der Lage Bakterien, Viren, Parasiten und giftige Schadstoffe zu bekämpfen und zu beseitigen. Von zentraler Bedeutung ist dabei die Unterscheidung zwischen körpereigen und körperfremd. Die beta-Aymloid-Plaques der Alzheimer-Krankheit enthalten als Hauptbestandteil beta-Amyloid-Peptide, Derivate aus dem körpereigenen beta-Amyloid-Vorläuferprotein, APP. Eine Impfung gegen beta-Amyloid erscheint daher zunächst widersinnig. Der Aggregationsvorgang jedoch, der über lösliche beta-Amyloid Peptide und diverse Zwischenstufen, wie z. B. Oligomere und Protofibrillen, schliesslich zu ausgereiften beta-Amyloid-Plaques führt, ist mit der Bildung von neuen, fibrillären, unphysiologischen Strukturen verbunden. Dies führt zur Präsentation von Neo-Epitopen, die sich von physiologischen Epitopen der beta-Amyloid-Peptide unterscheiden und damit als Angriffspunkt für eine Immunisierung eignen könnten.
Passive und aktive Immuntherapie
Immunisierung gegen ein Antigen kann in aktiver und passiver Form erfolgen. Das Grundprinzip der aktiven Immunisierung beruht auf der Präsentation von beta-Amyloid oder Derivaten als Antigen in Verbindung mit einem immunogenen Adjuvans, zusammen die „Vakzine“. Nach Injektion der Vakzine erfolgt die Erkennung des Antigens als körperfremd mit nachfolgender humoraler (Bildung von Antikörpern durch B-Lymphozyten) und zellulärer (Bildung antigenspezifischer T-Zellen) Immunantwort. Bei der passiven Immunisierung erfolgt die Gabe von biotechnologisch hergestellten, meist „humanisierten“, monoklonalen Antikörpern gegen beta-Amyloid direkt durch Injektion z. B. in das Venenblut („intravenös“) oder in das Unterhautfettgewebe („subkutan“).
Therapieforschung in Modellsystemen
Zur Erforschung neuer Therapieansätze menschlicher Krankheiten werden häufig experimentelle Modelle eingesetzt, zum einen in vitro- (Reagenzglas)-Modelle, zum anderen in-vivo-Modelle (z. B. transgene Fliegen oder Mäuse). In einem transgenen Maus-Modell der Alzheimer-Krankheit wurde 1999 erstmals eine Immunisierungstherapie mit mehrfachen Injektionen gegen beta-Amyloid versucht, mit überraschendem Erfolg: Die Impfung führte bei den geimpften Mäusen zu einer Immunreaktion mit Bildung von Antikörpern (körpereigene Abwehrstoffe) gegen beta-Amyloid. Als Folge konnten ein Abbau der beta-Amyloid-Plaques sowie eine Remission der Lernstörungen gemessen werden. Es zeigte sich dabei, dass die Bildung von Antikörpern nach der Impfung der entscheidende Schritt zum Therapieerfolg war.
Die ersten klinischen Studien: problematische Nebenwirkungen
Bei ersten klinischen Versuchen an Patienten traten mit diesem Ansatz jedoch bei 6% der Studienpatienten erhebliche Nebenwirkungen in Form von aseptischen Hirnentzündungen auf, so dass die Impfungen abgebrochen werden mussten. Klinische und neuropathologische follow-up Untersuchungen der immunisierten Patienten bestätigten einerseits die entzündlichen Nebenwirkungen, wahrscheinlich Ausdruck einer überschiessenden zellulären Immunantwort. Anderseits ergaben sich jedoch wichtige Hinweise auf die gezielte Bildung von Antikörpern gegen beta-Amyloid, der Übertritt von solchen Antikörpern über die Bluthirnschranke, die Entfernung von Amyloid-Plaques aus dem Gehirn und eine Verbesserung der Gedächtnisleistungen sowie eine klinische Stabilisierung bei Patienten, die Antikörper gebildet hatten.
Weiterentwicklung der Immuntherapie
Derzeit stehen in der Forschung Bemühungen in Vordergrund, den Angriffspunkt der Antikörper genauer zu definieren, die strukturellen Neoepitope gezielter zu erfassen und Autoimmunreaktionen besser auszuschliessen. Präklinische Versuchsreihen werden so gestaltet, dass das Risiko für Entzündungsreaktionen, Autoimmunkrankheiten und Blutungen minimiert werden kann. Es ist eine hohe Aktivität klinischer Studienprogramme zu verzeichnen, darunter überwiegend passive Immunsierungsansätze, aber auch aktive Impfungen. Die Entwicklungsstadien der klinischen Programme reichen (zum jetzigen Zeitpunkt: Oktober 2008) von Phase I (z. B. CAD106, Novartis; R1450, Roche; PF04360365, Pfizer; V950, Merck; GSK933776A, GlaxoSmithKline), über Phase II (z. B. LY2062430, Eli Lilly; IVIg–mix/Gammaguard, Baxter/Cornell) bis zur Phase III (Bapineuzumab, Elan/Wyeth). Ziel der Programme ist der Nachweis signifikanter klinischer Wirksamkeit bei gleichzeitiger ausreichender Sicherheit und Verträglichkeit der Immuntherapie.
Im Rahmen des Deutschen Stiftungstages 2016 in Leipzig hat Roland Bergfeld, Vorstandsvorsitzender Hans und Ilse Breuer-Stiftung, an einem Expertengespräch über Alterserkrankungen des Gehirns teilgenommen. Er sowie Prof. Dr. med. Thomas Gasser, Vorstandsvorsitzender Hertie-Institut für Hirnforschung und Direktor für Neurologie Universität Tübingen, Monika Kaus, Vorstandsvorsitzende der Deutsche Alzheimer Gesellschaft e.V. und Prof. Dr. Dr. h. c. Andreas Kruse, Direktor Institut für Gerontologie Universität Heidelberg, gaben in der von Julia Riedel (Hertie-Stiftung) moderierten Runde Einblicke über Herausforderungen und Perspektiven aus ihrem jeweiligen Kontext.
Es wurde deutlich, dass unsere Gesellschaft in vielen Bereichen auf einem guten Weg ist, aber sehr viel zu tun ist und wie eng die Anforderungen zusammenhängen.
By loading this video, you agree to the privacy policy of Youtube.